Gene regulatory network inference from CRISPR perturbations in primary CD4+ T cells elucidates the genomic basis of immune disease
Abstract
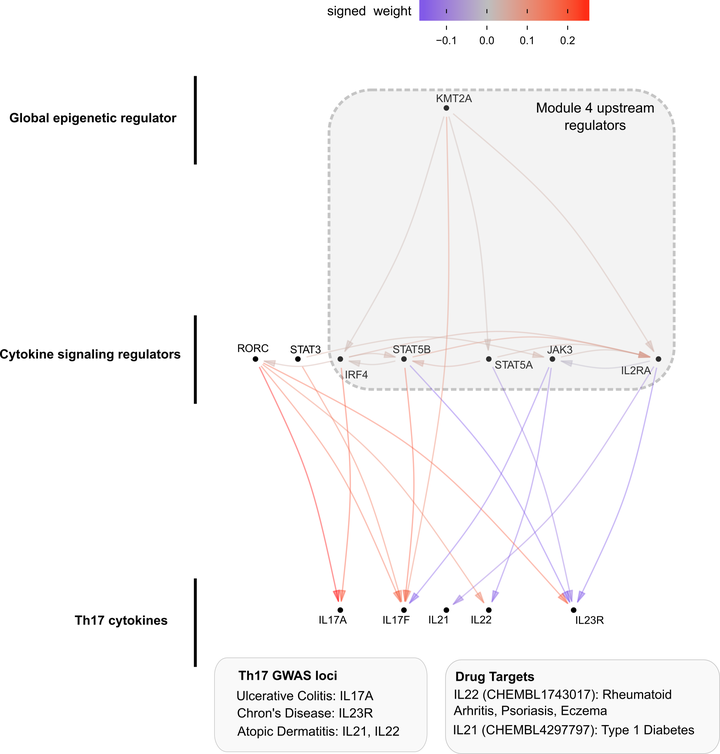
The effects of genetic variation on complex traits act mainly through changes in gene regulation. Although many genetic variants have been linked to target genes in cis, the trans-regulatory cascade mediating their effects remains largely uncharacterized. Mapping trans-regulators based on natural genetic variation, including eQTL mapping, has been challenging due to small effects. Experimental perturbation approaches offer a complementary and powerful approach to mapping trans-regulators. We used CRISPR knockouts of 84 genes in primary CD4+ T cells to perturb an immune cell gene network, targeting both inborn error of immunity (IEI) disease transcription factors (TFs) and background TFs matched in constraint and expression level, but without a known immune disease association. We developed a novel Bayesian structure learning method called Linear Latent Causal Bayes (LLCB) to estimate the gene regulatory network from perturbation data and observed 211 directed edges among the genes which could not be detected in existing CD4+ trans-eQTL data. We used LLCB to characterize the differences between the IEI and background TFs, finding that the gene groups were highly interconnected, but that IEI TFs were much more likely to regulate immune cell specific pathways and immune GWAS genes. We further characterized nine coherent gene programs based on downstream effects of the TFs and linked these modules to regulation of GWAS genes, finding that canonical JAK-STAT family members are regulated by KMT2A, a global epigenetic regulator. These analyses reveal the trans-regulatory cascade from upstream epigenetic regulator to intermediate TFs to downstream effector cytokines and elucidate the logic linking immune GWAS genes to key signaling pathways.